


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 synchrotron radiation photoelectron spectroscopy study of the oxidation of the Ge(100)-2
synchrotron radiation photoelectron spectroscopy study of the oxidation of the Ge(100)-2 1 surface by supersonic molecular oxygen beams
1 surface by supersonic molecular oxygen beams吉越 章隆; 寺岡 有殿; 岡田 隆太; 山田 洋一*; 佐々木 正洋*
Journal of Chemical Physics, 141(17), p.174708_1 - 174708_7, 2014/11
被引用回数:8 パーセンタイル:29.6(Chemistry, Physical)酸素分子の並進エネルギーを2.2eVまで変えた時のGe(100)21表面の飽和酸化まで表面状態をその場放射光光電子で調べた。飽和吸着酸素量が1モノレイヤー以下であり、Si表面酸化と大きく異なり酸化数が+2までであることが分かった。直接活性化吸着によるGe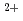 成分の増加を伴う吸着量の促進を観測した。本研究は室温における酸素吸着プロセスの基礎的理解に貢献する。
成分の増加を伴う吸着量の促進を観測した。本研究は室温における酸素吸着プロセスの基礎的理解に貢献する。
 D)+N
D)+N O
O NO+NO/N+O reaction
NO+NO/N+O reaction高柳 敏幸
Chemical Physics, 308(3), p.211 - 216, 2005/01
被引用回数:6 パーセンタイル:20.35(Chemistry, Physical)O(D)+NO反応について量子-古典波束法を用いた理論計算を行った。計算は平面対称性を仮定した5次元で行い、NO分子の3振動自由度を量子波束法によって取り扱い、残りの2自由度を古典力学で取り扱った。以前われわれが開発した高精度分子軌道法の計算結果をもとにして開発した解析的なポテンシャルエネルギー曲面を用いた。この計算の目的は2つの反応生成チャンネル、NO+NO及びN+Oが衝突エネルギーやNO分子の初期振動量子状態によってどのように変化するかを理論的に調べることである。計算の結果、衝突エネルギーの増加とともに、NO+NOの生成確率が減少し、N+Oチャンネルが逆に増加することを見いだした。一方、生成分岐比はNOの初期振動量子状態によってほとんど影響を受けないことがわかった。これらの計算結果は、成層圏での熱非平衡下で起こるO(D)+NO反応のメカニズムを理解するうえで極めて重要である。
D)+NONO+NO河合 信之輔*; 藤村 陽*; 梶本 興亜*; 高柳 敏幸
Journal of Chemical Physics, 120(14), p.6430 - 6438, 2004/04
被引用回数:8 パーセンタイル:25.15(Chemistry, Physical)O(D)+NO反応で生成するNO(v=0,1,2)の回転状態の分布を測定した。回転温度はおよそ20000Kであり、分布は位相空間理論で予想されるものに近いことがわかった。この結果は、反応中間体の寿命がそれほど長くはないが、分布はほぼ統計的であることを意味する。しかしながら、回転量子数の大きな場合には、分布は位相空間理論で予想されるよりも早く減衰した。このことを理解するため、分子軌道計算に基づいたポテンシャル曲面を用いて古典軌道計算を行った。その結果、実験で得られた高い回転量子数の分布が反応出口領域のポテンシャルの影響を強く受けることがわかった。
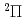 )+N(X
)+N(X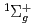 )
)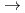 HCN(X
HCN(X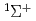 )+N(
)+N(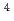 S) reaction
S) reaction和田 晃; 高柳 敏幸
Journal of Chemical Physics, 116(16), p.7065 - 7072, 2002/04
被引用回数:10 パーセンタイル:30.24(Chemistry, Physical)スピン禁制反応 CH(X)+N(X)HCN(X)+N(S) について、量子散乱理論を用いた計算を行った。CH分子を一個の原子とみなすことによって、自由度を3次元に落とした。分子軌道計算を用いて、スピン2重項及び4重項それぞれのポテンシャルエネルギー曲面を作製した。また、スピン軌道相互作用については過去の理論計算を用いた。超球座標を用いた堅密結合方程式を数値的に解いて、総反応確率を計算した。計算された確率は典型的な共鳴構造を示した。得られた確率から反応速度定数を計算し、実験結果と比較したところ、100倍ほど小さな値が得られたが、速度定数はスピン軌道相互作用に大きく依存することがわかった。
 anion
anion高柳 敏幸; 和田 晃
Chemical Physics Letters, 348(1-2), p.514 - 520, 2001/11
被引用回数:14 パーセンタイル:41.25(Chemistry, Physical)時間に依存しない量子反応性散乱理論を用いてF(HD)アニオンの光電子脱離スペクトルの計算を行った。StarkとWernerの作製した高精度のポテンシャル面を使った。計算したFHD及びFDH両アニオンのスペクトルには、束縛回転に相当するブロードなピークがいくつか見られた。これは、以前研究されたFH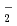 のスペクトルで見られたものと本質的に同じである。さらに、FHDアニオンでは、遷移状態共鳴に相当するピークが見られた。これは、最近、詳細な反応断面積の測定によって実験的に見出されているものである。本理論計算結果は光電子脱離スペクトル実験によって、遷移状態共鳴が見出される可能性があることを強く示唆するものである。
のスペクトルで見られたものと本質的に同じである。さらに、FHDアニオンでは、遷移状態共鳴に相当するピークが見られた。これは、最近、詳細な反応断面積の測定によって実験的に見出されているものである。本理論計算結果は光電子脱離スペクトル実験によって、遷移状態共鳴が見出される可能性があることを強く示唆するものである。
D)+NONO+NO reaction高柳 敏幸; 和田 晃
Chemical Physics, 269(1-3), p.37 - 47, 2001/07
被引用回数:14 パーセンタイル:41.25(Chemistry, Physical)O(D)+NONO+NO反応について、量子反応性散乱計算を行った。ポテンシャルエネルギー曲面は、CASPT2レベルの高精度の分子軌道計算を行い、解析関数にフィットして作製した。反応側及び生成側の配向角を固定したモデルを用いることによって、次元を3次元に落とした。この反応では2種類のNO分子が生成する。反応熱はおもに新しく生成するNO分子の振動に分配されるが、もともと存在したNO振動モードにも、ある程度エネルギーが分配されることを見出した。このことはもともと存在したNO結合が、必ずしもスペクテータではないことを示している。
 HF and HDF van der Waals molecules
HF and HDF van der Waals molecules高柳 敏幸; 和田 晃
Chemical Physics Letters, 338(2-3), p.195 - 200, 2001/04
被引用回数:15 パーセンタイル:43.4(Chemistry, Physical)ファン・デル・ワールス分子の前期反応過程,D…HF+h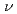 →DH+F及びH…DF+h→HD+Fについて理論的に検討した。超球座標を用いた3次元の時間に依存しない量子反応性散乱理論を使って計算を行った。また正確な分子軌道計算によって作成されたポテンシャルエネルギー曲面を用いている。その結果、水素原子移動反応であるD…HF+h→DH+F過程は非常に大きな確率で起こるが重水素原子の移行するH…DF+h→HD+F反応の確率は極めて小さいことがわかった。このことは前期反応過程ではトンネル効果が支配的な役割をすることを示している。
→DH+F及びH…DF+h→HD+Fについて理論的に検討した。超球座標を用いた3次元の時間に依存しない量子反応性散乱理論を使って計算を行った。また正確な分子軌道計算によって作成されたポテンシャルエネルギー曲面を用いている。その結果、水素原子移動反応であるD…HF+h→DH+F過程は非常に大きな確率で起こるが重水素原子の移行するH…DF+h→HD+F反応の確率は極めて小さいことがわかった。このことは前期反応過程ではトンネル効果が支配的な役割をすることを示している。
寺岡 有殿; 吉越 章隆
Applied Surface Science, 169-170, p.738 - 741, 2001/01
被引用回数:68 パーセンタイル:91.83(Chemistry, Physical)SPring-8に建設された原研軟X線ビームラインBL23SUに設置される表面反応分析装置(エンドステーション)の設計及び基本的な性能について現状を報告する。本装置は主に表面反応分析室、表面構造分析室、ビームモニタ室、超音波分子線発生器から構成される。表面反応分析室では電子エネルギー分析と質量分析の予備実験結果を紹介する。表面構造分析室ではサンプルのクリーニングとその後の表面分析(LEED/AES)について述べる。ビームモニタ室については差動排気の達成度を報告する。超音速分子線発生器ではN及びO分子線を発生させ、差動排気の達成度や化学組成の質量分析結果を報告する。本報告では以上の基本的な性能が表面反応ダイナミクスの研究に不可欠であることを強調する。
/Si(001)系における初期反応ダイナミクス寺岡 有殿; 吉越 章隆
日本真空協会関東支部平成13年度第3回研究例会資料, p.1 - 8, 2001/00
BL23SUに設置した表面反応分析装置を用いて、超音速O分子線を使って形成したシリコン酸化膜を高分解能放射光を活用してその場光電子分光測定し、清浄Si(001)表面と残留HOにより部分的に酸化されたSi(001)表面の初期酸化に対するO分子の並進運動エネルギーの影響を3eVまでの範囲で実験的に研究した。部分酸化表面で明らかに見られた閾値は清浄表面では不明瞭であった。その原因は清浄表面では二量体の架橋位置からO原子がバックボンドに室温でも自己拡散するのに対して、部分酸化表面ではダングリングボンドを終端したH原子やOH基が拡散経路を阻むためと解釈された。Si-2pや光電子分光から二量体にHOが解離吸着した部位はO分子によって酸化されにくいにもかかわらず、運動エネルギーの作用で酸化が促進され室温では0.5nm程度まで極薄酸化膜形成を制御できることが明らかとなった。
 P
P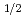 )+H HBr+H reaction
)+H HBr+H reaction高柳 敏幸; 黒崎 譲
Journal of Chemical Physics, 113(17), p.7158 - 7164, 2000/11
被引用回数:43 パーセンタイル:77.61(Chemistry, Physical)スピン軌道相互作用による電子的非断熱遷移を伴う反応、Br(P)+H HBr+Hについて3次元量子反応性散乱計算を2つの計算方法を用いて行った。1つは超球座標を用いたclose-coupling法で、もう一方は、虚数の吸収ポテンシャルを用いた一般化R行列伝播法である。後者では反応側のJacobi座標を用いた。ポテンシャル曲面としてはTruhlarらによる(22)のdiabaticなポテンシャル曲面を用いた。いずれの方法でも数値的に十分収束した計算結果を得ることができた。また、得られた結果から電子的非断熱遷移が反応の入口でほとんど起こるが、その効率は小さいことがわかった。
D)+CH reaction; Does the N(D) atom really insert into CH bonds of alkane molecules?
reaction; Does the N(D) atom really insert into CH bonds of alkane molecules?高柳 敏幸; 黒崎 譲; 横山 啓一
International Journal of Quantum Chemistry, 79(3), p.190 - 197, 2000/09
被引用回数:11 パーセンタイル:49.22(Chemistry, Physical)最近、米国の量子化学研究者によってN(D)原子がメタンのCH結合に挿入しないことが報告されたが、本論文はその研究結果に対する反論である。多配置ハートリーフォック計算、さらに大規模な配置間相互作用を考慮した計算によって、N(D)原子がCH結合に挿入してCH NH(A'')を生成することを改めて理論的に示した。さらに興味深いことに二重項第一励起状態のポテンシャル曲面上でも挿入反応が起こることを見いだした。この場合はCHNH(A')分子が生成する。これらの結果は最近われわれが行ったN(D)+Hのポテンシャル曲面の結果とよく似ている。
NH(A'')を生成することを改めて理論的に示した。さらに興味深いことに二重項第一励起状態のポテンシャル曲面上でも挿入反応が起こることを見いだした。この場合はCHNH(A')分子が生成する。これらの結果は最近われわれが行ったN(D)+Hのポテンシャル曲面の結果とよく似ている。
Balucani, N.*; Algia, M.*; Cartechini, L.*; Casavecchia, P.*; Volpi, G. G.*; 佐藤 圭*; 高柳 敏幸; 黒崎 譲*
Journal of the American Chemical Society, 122(18), p.4443 - 4450, 2000/05
被引用回数:77 パーセンタイル:87.92(Chemistry, Multidisciplinary)第一励起状態であるN(D)原子のアセチレンとの反応について、公差分子線と高いレベルの分子軌道計算によって調べた。主たる反応メカニズムはN(D)+CHHCCN+Hであり、窒素と水素原子が交換する。この反応はタイタンの大気化学に非常に重要であることが予想される。これまで大気中のCHを含んだ化合物はほとんどイオン分子反応で生成すると考えられていたが、本研究は中性分子間の反応も重要であることを示す。
S, D, P)+H reactions高柳 敏幸; 黒崎 譲; 横山 啓一
Chemical Physics Letters, 321(1-2), p.106 - 112, 2000/04
被引用回数:24 パーセンタイル:58.71(Chemistry, Physical)多配置参照配置間相互作用の方法を用いた分子軌道法によってN(S, D, P)+Hの反応のポテンシャルエネルギー曲面を計算した。特にC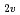 とCoor配置の計算を重点的に行い、2次元のポテンルシャル面を詳細に作製した。その結果、N(D)+H反応については5枚のポテンシャル面のうち、2枚が重要であることを明らかにした。またこれらのポテンシャル面が最低4重項のポテンシャルと交差し、N(S)+Hへの非断熱遷移が起こりうることを見いだした。また計算結果に基づき、N(P)+Hの消光過程のメカニズムについて検討した。
とCoor配置の計算を重点的に行い、2次元のポテンルシャル面を詳細に作製した。その結果、N(D)+H反応については5枚のポテンシャル面のうち、2枚が重要であることを明らかにした。またこれらのポテンシャル面が最低4重項のポテンシャルと交差し、N(S)+Hへの非断熱遷移が起こりうることを見いだした。また計算結果に基づき、N(P)+Hの消光過程のメカニズムについて検討した。
) reaction system
reaction system高柳 敏幸; 黒崎 譲; 市原 晃
Journal of Chemical Physics, 112(6), p.2615 - 2622, 2000/02
被引用回数:62 パーセンタイル:86.41(Chemistry, Physical)非断熱遷移を伴う(D+H)イオン分子反応について3次元量子散乱計算を行った。超球座標を使った時間に依存しないclose-coupling法を用いた。ポテンシャルエネルギー曲面として(33)のDIMポテンシャルを使った。正確な量子論の計算結果を半古典的なトラジェクトリホッピングの結果と比較した。その結果Tullyによって提唱されている方法のほうが従来から使われているTully-Prestonの方法よりも量子論の結果をよく再現することがわかった。これはTully-Prestonの方法が、ポテンシャルの交差付近でのみの電子遷移しか考慮していないことが原因である。
…HD van der Waals molecule高柳 敏幸; 黒崎 譲
Physical Chemistry Chemical Physics, 2(4), p.665 - 670, 2000/02
被引用回数:10 パーセンタイル:30.33(Chemistry, Physical)赤外励起によって引き起こされるファン・デル・ワールス分子の前期反応過程、H…HD+hH+Dについて反応性散乱理論を使った理論的研究を行った。正確な分子軌道計算をもとにして作製されたStarck-Meyerのポテンシャルエネルギー曲面を用いた。その結果H…HD(=1)という共鳴状態を経ると、反応が5 10%の確率で起こることが予想された。また回転励起に関する共鳴状態を経由した場合、ほとんど前期解離過程H…HD+hH+HDが起こることがわかった。この結果はH
10%の確率で起こることが予想された。また回転励起に関する共鳴状態を経由した場合、ほとんど前期解離過程H…HD+hH+HDが起こることがわかった。この結果はH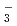 ポテンシャル曲面においては回転非断熱遷移の確率が大きいことを意味している。
ポテンシャル曲面においては回転非断熱遷移の確率が大きいことを意味している。
D)+acetylene reaction高柳 敏幸; 黒崎 譲*; 横山 啓一; 佐藤 圭*; 綱島 滋*
Chemical Physics Letters, 312(5-6), p.503 - 510, 1999/00
被引用回数:9 パーセンタイル:28.09(Chemistry, Physical)分子軌道計算結果を用いて、N(D)+CH,CD反応の反応速度定数の変分的遷移状態理論計算を行った。計算値と実験値の比較から、量子力学的効果である非断熱過程が重要であることを明らかにした。さらにこのことを半定量的に確かめるために、正確な分子軌道理論を用いて、長距離領域でのポテンシャルエネルギー曲面の計算を行った。その結果ファンデルワールス領域でポテンシャルの交差が起こっていることがわかった。
D) with CHOH and its isotopomers梅本 宏信*; 金剛 晃一*; 稲葉 重信*; 園田 保之*; 高柳 敏幸; 黒崎 譲
Journal of Physical Chemistry A, 103(35), p.7026 - 7031, 1999/00
被引用回数:8 パーセンタイル:25.43(Chemistry, Physical)メタノールとN(D)原子の反応経路をレーザー誘起けい光法とab initio分子軌道法を用いて調べた。NH及びOHラジカルが反応生成物として検出され、これらの分子の内部状態が統計的でないことが確認された。このことは反応中間体の寿命がエネルギーがランダムになる時間より短いことを示している。CHOD及びCDOHとの反応からNH,ND,OH及びODが生成物として検出された。測定された生成物の分岐比から、N原子はCH結合に挿入しその後水素原子のスクランブリングが起こると結論された。この結果は分子軌道法の計算結果と定性的に一致することがわかった。
F molecule高柳 敏幸; 黒崎 譲*; 田池川 浩人*
Int. J. Mass Spectrom. Ion Process, 176(3), p.227 - 235, 1998/00
被引用回数:4 パーセンタイル:21.15(Physics, Atomic, Molecular & Chemical)CClF分子のイオン化及び電子付着過程に引き続いて起こる分子のダイナミクスについての情報を得るためにab initioダイナミクス計算を行った。垂直イオン化によって生成するCClFは非常に短い時間でCClFとClに解離する。この時、80%のエネルギーには2つのフラグメントの並進運動エネルギーに変換されることがわかった。一方、垂直電動エネルギーによって生成するCClFアニオンも非常に短い時間内でCClFとClに解離する。しかしこの場合、エネルギーの大部分はCClFの内部エネルギーに変換する。カチオンとアニオンの解離ダイナミクスの違いについて、ポテンシャルエネルギー曲面の違いを基に議論した。
D,P)+CH and CD reactions高柳 敏幸; 黒崎 譲*; 三沢 和秋*; 杉浦 円*; 小林 康英*; 佐藤 圭*; 綱島 滋*
Journal of Physical Chemistry A, 102(31), p.6251 - 6258, 1998/00
被引用回数:42 パーセンタイル:78.2(Chemistry, Physical)パルスラジオリシス共鳴吸収法を用いてN(D,P)とCH及びCDの反応速度定数の測定を行った。反応速度定数の温度依存からアレニウスパラメータを決定した。得られた活性化エネルギーはN(D)で約0.5kcalmol 、N(P)で約0.9kcalmolであった。H/Dの同位体効果はN(D),N(P)とも非常に小さいことがわかった。また反応速度定数の絶対値についてはN(D)のほうが約3倍大きい。N(D)+CHの反応のメカニズムを調べる目的で、ab initio分子軌道計算を行った。それによるとN(D)はCHの
、N(P)で約0.9kcalmolであった。H/Dの同位体効果はN(D),N(P)とも非常に小さいことがわかった。また反応速度定数の絶対値についてはN(D)のほうが約3倍大きい。N(D)+CHの反応のメカニズムを調べる目的で、ab initio分子軌道計算を行った。それによるとN(D)はCHの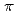 結合に付加することがわかった。実験で得られた反応速度定数と遷移状態理論による速度定数の比較を行い、理論計算の結果の妥当性について論じた。
結合に付加することがわかった。実験で得られた反応速度定数と遷移状態理論による速度定数の比較を行い、理論計算の結果の妥当性について論じた。
H+CH reaction高柳 敏幸
Journal of Chemical Physics, 104(6), p.2237 - 2242, 1996/02
被引用回数:97 パーセンタイル:94.08(Chemistry, Physical)H+CHH+CH反応について次元を落とした量子反応性散乱理論を用いて調べた。系を直線4原子の反応として取り扱い、数学的な次元を3次元にまで少なくした。振動モードとしては、CHの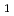 およびモード、Hの振動、CHのかさ振動であるモードが考慮された。ポテンシャルエネルギー曲面としてはJordanとGilbertによる半経験的ポテンシャル関数を用いた。回転平均した反応断面積および反応速度定数はエネルギーシフト近似を用いて計算した。計算の結果、CHのモードの励起が反応性に著しく影響を与えることがわかった。これは反応座標とモードのカップリングが強いことを示している。またHとCHの振動分布について調べたところ、Hはあまり振動励起していないが、CHのモードは反応によって励起していることがわかった。
およびモード、Hの振動、CHのかさ振動であるモードが考慮された。ポテンシャルエネルギー曲面としてはJordanとGilbertによる半経験的ポテンシャル関数を用いた。回転平均した反応断面積および反応速度定数はエネルギーシフト近似を用いて計算した。計算の結果、CHのモードの励起が反応性に著しく影響を与えることがわかった。これは反応座標とモードのカップリングが強いことを示している。またHとCHの振動分布について調べたところ、Hはあまり振動励起していないが、CHのモードは反応によって励起していることがわかった。
